
~ Methods ~
Clean and marker-free deletion with the ∆manP-manA system
Recently, a new method that allows base pair-precise and marker-free deletions in B. subtilis has been developed (Wenzel et al., 2015). This method uses an integrative plasmid that confers spectinomycin resistance and introduces manP, coding for the mannose-specific EIIBCA of the phosphotransferase system (Fig. 1A and 1B). In the B. subtilis wild-type, mannose is taken up and phosphorylated simultaneously by ManP. Subsequently, the produced mannose 6-phosphate gets isomerized to fructose 6-phosphate by the mannose 6-phosphate isomerase encoded by manA. However, if manA is absent and manP is present, mannose-6-phosphate accumulates and becomes toxic for the cell.
In order to perform a deletion, manPA-deficient strains are transformed with deletion plasmids and cells with integrated plasmids are selected by spectinomycin. The deletion of the desired genomic region can occur when the additional homologues regions recombine. When adding mannose to the medium, it is possible to select for the loss of the counter-seletion marker manP, and thus, for the deletion. Since, the plasmid can recombine by itself, PCRs have to be performed to check the correct deletion (Fig. 1B and 1C).
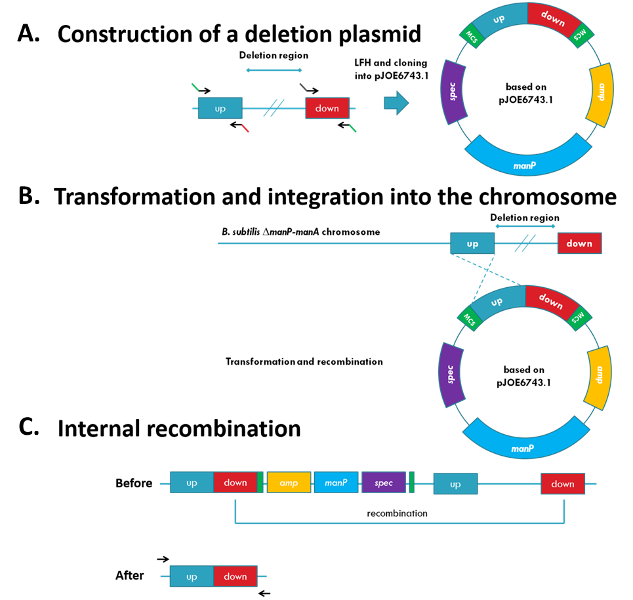
Figure 1: Clean and marker-free deletion
(A) The single parts of the deletion plasmids are amplified by PCR and joined with a LFH-PCR. The product is cloned into pJOE6743.1 (B) The plasmid can integrate by the ‘up’ or ‘down’ region into the chromosome by homologous recombination. (C) The deletion of the desired genomic region occurs when the additional homologues regions recombine. The successful deletion is checked by PCR.
Gibson DNA-assembly
The Gibson assembly is an easy way to construct artificial operons, plasmids or even complete chromosomes (Gibson et al., 2009). For this purpose, the DNA fragments need to share homologous sequences at their 3’- and 5’-ends, respectively. The fragments are incubated together with the T5 exonuclease, a polymerase and a thermostable ligase at 50°C. The T5 exonuclease causes a “chew back” of the DNA fragments allowing them to anneal to each other. The exonuclease is thermolabile, thus it is active only for the first few minutes. After the single-stranded DNA molecules have annealed, the polymerase re-synthesizes the strands, while the ligase combines them to one double-stranded DNA fragment (Fig. 22). This method has been shown to function with up two 4 fragments at the same time.
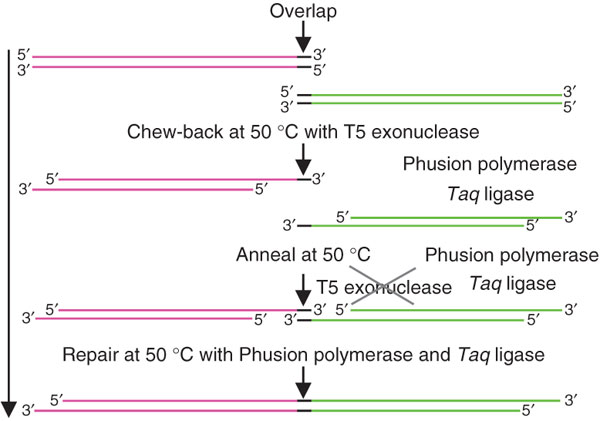
Figure 2: DNA assembly with the Gibson DNA assembly
The dsDNA fragments to be combined share overlapping, homologous 3’- and 5’-end. The added T5 exonuclease is responsible for the “chew back” of the dsDNA. This allows the strands to anneal. The Phusion polymerase re-synthesizes the strands and the Taq ligase combines them to one single dsDNA fragment (picture adopted from Gibson et al., 2009).
Ligase cycling reaction (LCR)
This method allows scarless assembly of up to 20 DNA fragments in a single step (de Kok et al., 2014). For this purpose, double-stranded DNA-fragments are mixed with single-stranded bridging primers. These primers have homologous parts for each double-stranded DNA part A and B (Fig.3). As in a standard PCR, the dsDNA is denaturated at 94°C, and the ssDNA bridging primer is allowed to anneal during cooling down. A thermostabile ligase recognizes the gap between the fragments A and B and combines the strands at 66°C. The repetition of these steps results in the assembly of all added fragments. The method has been shown to be even more precise than the Gibson assembly that causes SNPs and insertions/deletions with a frequency of about 1 per 25 and 50 kb, respectively. In our project this method will be the first choice for the assembly of artificial operons in order to increase the work flow.
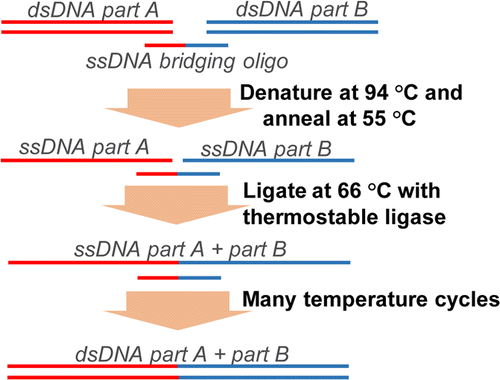
Figure 3: DNA assembly by Ligase cycling reaction
The LCR combines up to 20 DNA fragments scarless in a cycling reaction. For this purpose, dsDNA fragments (A and B) are mixed with a ssDNA bridging primer. The primer is specific for the 3’- and 5’-ends of the respective DNA fragments. As in a normal PCR the mixture undergoes heating and cooling steps, so the dsDNA can denaturate, the primer can bind and the ligase can combine the ssDNA fragments. By repeating these steps several times all added dsDNA fragments are combined (picture adopted from de Kok et al., 2014).